Information about new Medical Device Regulation (MDR) for Fresenius Kabi's products
Fresenius Kabi is a manufacturer of a large portfolio of medical devices covering several therapeutic fields. Therefore, Fresenius Kabi is widely impacted by the new Medical Device Regulation (MDR) EU 2017/745 which becomes valid from May 2021 onwards. Many questions arise concerning the MDR regarding the impact on the availability of Fresenius Kabi products and the general preparation and adaptation status.
MDR impacts all Medical Devices, from class I to class III. There are more and stricter requirements than with the Medical Devices Directive in the past.
Due to the extent and complexity of the new MDR and in order to be able to guarantee a continuous supply of our medical devices, Fresenius Kabi started at an earlier point in time to implement dedicated programs on several levels of our organization. At all levels the work and progress are duly monitored and in close coordination with our notified bodies.
In summary, we can say that Fresenius Kabi has changed the way we were working in the past to become more clinical oriented, based on the post market input. The aim is to confirm the safety and performance throughout the expected lifetime of the device and to guarantee the continued acceptability of identified risks by detecting the emerging risks based on factual evidence. The following Q&A section will provide the main information on the MDR implementation by Fresenius Kabi, and the consequences for our customers and users of our medical devices.
Questions & Answers
How does Fresenius Kabi AG handle the transfer from MDD to MDR?
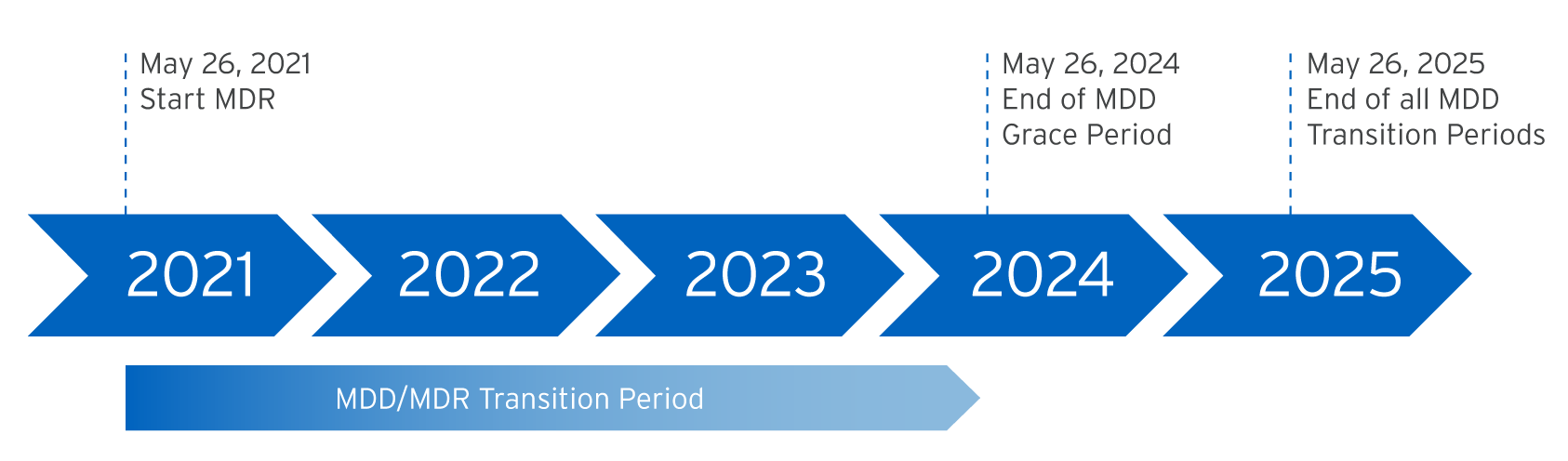
As of May 26, 2021, the EU legislation is based on the Medical Device Regulation (EU) 2017/745.
Currently the basis for the product conformity is - beside the new MDR - the Medical Device Directive 93/42/EEC (MDD) and this will be valid for unchanged products until the end of the validity date, given our EC certificate (May 2024).
This measure means we will have certain products on the market that still refer to the old MDD while others will have changed to the new MDR. The facts will be reflected in the declarations of conformity.
In the meantime, Fresenius Kabi will place both MDD and MDR certified medical devices on the market and, until May 2024, Fresenius Kabi products with MDD certification will gradually be certified according to MDR. Within this context we work in a very close co-operation with our Notify Bodies, which is the only way to guarantee a smooth and controlled transition at any time.
Until May 2025, a sell-off of stocked MDD certified products may still take place, which will be restocked with MDR certified products.
What are the actions and challenges to fulfill the new MDR requirements at Fresenius Kabi AG?
Since 2017 Fresenius Kabi has established a transition plan and a special team is dedicated to working in the adaptation to the new medical device legislation. The MDR brought a lot of highly demanding and very cost intensive new tasks. Therefore, Fresenius Kabi adapted several internal processes and increased in several fields the manpower to be able to fulfil all new requirements.
Our processes have changed to adapt each technical dossier that has taken between 4 and 6 months to build them according to the new requirements.
In details major adaptations were needed and have been achieved in the field of:
- General Quality Management System and process definition and structure
- Labelling, instructions for use, and UDI
- Clinical Evaluation
- Risk Management
- Usability
- Post Market Surveillance, Post Market Clinical Follow-up
- Distributor obligations
- Fulfilment of new “General Safety and Performance Requirements”
- EU Declaration of Conformity
- Fulfilment of “state of art”
- Technical file structure
How does Fresenius Kabi AG handle Post market surveillance?
Another major change in the processes of Fresenius Kabi has been the reinforcement of the post-market surveillance process. Manufacturers of Medical Devices shall prepare this surveillance report summarizing the results and conclusions of the analysis of the data gathered from the post market surveillance activities.
The new MDR has many more requirements and is stricter than the MDD, therefore we had to respond to the clinical need with a new expert review. Several months were needed to create these new documents for all departments to validate it.
Post-market surveillance (PMS) is defined as a "systematic process to derive necessary corrective and preventive actions (CAPA) from information on medical devices already placed on the market". To face the difficulty of assessing all risks considered only during the development and production phase of a product, the Post-market surveillance aims at
- identifying risks when they occur during practical usage
- monitoring of product performance in the field
- detecting any failure in the product while used in the field
PMS system uses several different sources of information to be able to generate all the frequent new reports which have been established in the MDR. Several new activities have been defined to get all necessary information in a proactive manner. One of these new activities is very crucial for the PMS and needs the active involvement of our customers. To get firsthand information about the usage and behavior of our products in the field we need to approach our customers and proactively ask them for their feedback. To reduce this burden for our customers as far as possible we have created an internet-based software tool which allows an anonymous distribution.
Fresenius Kabi is conscious that we are asking for an extra effort from our customers. Not only we are obliged by the MDR to conduct such surveillance activities, but this input will be our basis to lean on when searching for a larger clinical insight to build the future of our devices.
How does Fresenius Kabi AG implement UDI?
The Unique Device Identification (UDI) is a series of numeric or alphanumeric characters that is created through a globally accepted device identification and coding standard, enabling an unambiguous identification of a specific medical device on the market.
The UDI Carrier shall be on the label or on the device itself and on all higher levels of device packaging.
UDI is comprised of:
- UDI Device identifier (UDI-DI) and
- UDI Production identifier (UDI-PI).
UDI-DI is specific to a device, providing information about the product (article code) and the manufacturer.
Information like a lot number, serial number, software identification or expiry date appearing on the label shall be part of the UDI-PI.
It is important to note that no UDI-PI information can be included in the UDI database of EUDAMED but only UDI-DI information.
Timelines for the implementation of UDI on the product are as follows:
| Device as per MDR |
Implantable devices and Class III devices |
Class IIa and Class IIb devices |
Class I devices |
|---|---|---|---|
| Placing UDI-carriers on device labels (MDR Article 123 (3)f, Article 27(4) |
May 26, 2021 | May 26, 2023 | May 26, 2025 |
In contrast to the timelines given by the MDR Fresenius Kabi will also put UDI on its Class I products from May 2021 onwards.
Will there be changes to the labels and instructions for use due to the MDR?
We adapt all products from our portfolio: MDR certified products require a new label design as several new symbols have been introduced within this context. All MDR compliant products will have a symbol to indicate that the product is a medical device. MDR certified products also require a Unique Device Identifier (UDI) which is also indicated by a new symbol. Besides these two examples, further new symbols have been established. Examples are shown in the illustration.
Will products be discontinued due to MDR?
In general, there will be almost no product discontinuations due to the MDR. In the process, we have filtered some products which are substituted by newer versions and we changed the classification of some others in order to better adapt them to the new standards. New tests, new procedures, and new technical dossiers have been established to avoid blocking our customers.
How does Fresenius Kabi AG support its distributors to fulfil their legal requirements?
According to Article 14 of the new MDR, distributors of our products have the obligation to verify on a regular basis that several requirements for our products are met. These are - for instance - a proper CE marking, UDI, Instruction for use, etc.
To help our distributors to get access to this kind of information for their products, Fresenius Kabi has opened up a database available without access control on a dedicated website. All information is easily accessible through a search filter with a simple article code.